

Процесс восстановления озона в рекомбинационном процессе (1) является
одним из важных в атмосфере (рис. 1). Хорошо известно, что в этом процессе озон образуется колебательно возбужденным O3(υ).
Дальнейшая судьба молекулы озона O3(υ) решается в двух конкурирующих процессах. Часть молекул O3(υ) стабилизируется в столкновительных процессах (3) и
(4b) и радиационном процессе (6). Другая часть молекул O3(υ)
гибнет в ходе химического взаимодействия с активными компонентами
атмосферы, такими как атомом кислорода (процесс (4а)) и с
электронно-возбужденной молекулой кислорода O2(a1Δ) (процесс (2)). В качестве активных частиц могут выступать целый ряд других компонент атмосферы, такие как NO, NO2, OH и т.д. (процесс (5)).

Рис. 1.
Кинетическая схема показывающая формирование (formation), гибель
(destruction) и стабилизацию (stabilization) колебательно-возбужденного
озона в атмосфере.
Необходимо отметить, что термализованный озон O3 также вступает в реакции с активными компонентами атмосферы, но значительно с меньшей эффективностью, чем колебательно возбужденный O3(υ). Например, константа скорости процесса (2) на 4 порядка выше константы скорости процесса тушения O2(a1Δ) на термализованном озоне. Процессы (2), (4a) и (5) приводят к замедлению скорости восстановления озона и к увеличению скорости убыли молекул O2(a1Δ) и атомов кислорода O в смеси O/O2/O3. Это необходимо учитывать в расчетах концентрационных профилей O, O3 и O2(a1Δ) в атмосфере. Пренебрежение этими процессами может привести к значительным систематическим ошибкам при измерении кинетических констант атмосферных процессов в лабораторных условиях.
В настоящий момент имеется весьма скудная информация по процессам с участием активных форм кислорода. Это связано прежде всего со сложностью проведения экспериментальных исследований процессов в котором сталкивающиеся частицы являются радикальными. Целью наших исследований является измерение кинетических констант процессов с участием активных компонент атмосферы.
Мы проводим лабораторные исследования атмосферных процессов с участием молекулярного синглетного кислорода O2(a1Δ), атомарного кислорода в основном и первом возбужденном состоянии и колебательно-возбужденного озона. Рисунок 2 показывает общий вид лабораторной установки. Атомы кислорода и молекулы O2(a1Δ) нарабатываются УФ фотолизом озона. Источником УФ излучения служат эксимерный лазер (Lambda Physik Compex Pro 102, длительность импульса 10 нс, частота повторения импульсов до 10 Гц) или 4-ая гармоника от твердотельного лазера Nd:YAG (Solar Systems LQ829, длительность импульса 10 нс, частота повторения импульсов до 10 Гц, энергия в импульсе при 266 нм около 100 мДж).
Кинетика тушения O2(a1Δ) отслеживалась наблюдением за его люминесценцией на длине волны 1268 нм. Временные зависимости концентрации атомов кислорода регистрировались с использованием хемилюминесцентной реакции O+NO. Время-разрешенная абсорбционная спектроскопия использовалась для регистрации временных зависимостей озона в зоне фотолиза.

Рис. 2. Фотография общего вида фотохимической лаборатории.
Основные узлы экспериментальной установки показаны на фотографии 3. Фотолизная ячейка подробно описана в недавней работе (Azyazov V.N. et al. Chem. Phys. Lett., vol. 482, 2009, pp. 56-61, doi: 10.1016/j.cplett.2009.09.095).

Рис. 3. Фотография показывающая основные узлы установки. 1 – фотолизная ячейка, 2 – импульсная лазерная система, 3 – монохроматор, 4 - ФЭУ.
В ходе наших исследований мы обнаружили ряд новых эффектов в чистой кислородсодержащей среде:
1) Эффект быстрого тушения синглетного кислорода O2(a1Δ). Молекула O2(a1Δ) весьма устойчива к столкновениям. Вероятность ее дезактивации при столкновениях мизерная. Однако, Рахимова Т.В. и соавторы (МГУ, Институт ядерной физики им. Д.В. Скобельцина)) заметили быстрое падение концентрации синглетного кислорода в присутствии атомов кислорода в послеразрядной зоне. Наблюдаемая быстрая убыль O2(a1Δ) не могла быть объяснена тушением в известных бинарных процессах.
В своих экспериментах по импульсному лазерному фотолизу озона мы также обнаружили эффект быстрого тушения синглетного кислорода в кислородсодержащей среде. На рис. 4 представлены временные зависимости интенсивности люминесценции O2(a1Δ) полученные после ИЛФ на длине волны 248 нм при давлении озона PO3=1 Toрр, температуре смеси T=300 K, удельной энергии лазерного импульса E=87 мДж/cм2, давлении кислорода PO2 = 460 Toрр с разбавкой CO2 в интервале давлений 6.7-97 Toрр.

Рис. 4. Временные профили интенсивности люминесценции кислорода на длине волны 1268 нм I после ИЛФ смеси O3/O2/CO2 при E=87 мДж/cм2 и T=300 K.
Два временных масштаба на временных профилях интенсивности люминесценции O2(a1Δ) отчетливо видны. Наблюдается быстрый спад на первых 30 микросек. Затем наблюдается более пологий спад который объясняется тушением O2(a1Δ) на озоне. Быстрая дезактивация на первом участке нельзя было объяснить не одним из известных процессов. Мы предположили, что O2(a1Δ) в этой области тушится колебательно-возбужденным озоном. Действительно, добавки в смесь компонентов хорошо тушащих приводили к замедлению тушения O2(a1Δ). Как это видно из рис. 4 добавление CO2 резко подавляет процесс тушения.
2) Эффект неполного восстановления озона. Существующие кинетические схемы рекомбинации атомов кислорода в процессе (1) при большом избытке молекулярного кислорода предсказывают практически полную конвертацию атомов кислорода в молекулы озона. В экспериментах по УФ фотолизу озона мы обнаружили, что существенная доля разваленного в ходе фотолиза озона не восстанавливается до первоначального уровня. Рис. 5 показывает зависимости концентрации озона от времени после ИЛФ смеси O2/O3/CO2/Ar при E=70 мДж/cм2, полном давлении Ptot =712 Toрр, давлении кислорода PO2 =182 Toрр, T=300 K, начальной концентрации O3 N0O3=2.75x1016 cм-3. Давления Ptot и PO2 поддерживались постоянными. Давления Ar и CO2 изменялись так, чтобы PAr + PCO2= 530 Toрр. Видно что некоторая часть молекул озона диссоциирует дополнительно в химических процессах с участием атомов O(1D) на временном интервале < 5 микросек. CO2 хороший тушитель O(1D) поэтому его добавление подавляет этот канал диссоциации озона, как это видно из рис. 5.
Результаты
представленные на рис. 5 показывают что степень восстановления озона в
некоторых экспериментах далеко не доходит до 100 % уровня. Скорости
удаления озона в известных реакциях
O3 + O(3P) → O2
+ O2
(8)
O + O + M → O2
+ M
(9)
много
меньше скорости рекомбинационного процесса (1), поэтому они дают
незначительный вклад в убыль молекул озона. Так например, для смеси O2/O3/Ar (нижняя кривая на рис. 5) степень восстановления озона составляет только около 70 %. Опять же добавление CO2
путем замены Ar
вызывает заметное увеличение степени восстановления озона. Данные
результаты свидетельствуют о важности процесса (4а) в
кинетике озона.

Рис. 5. Временные профили концентраций O3 при E=70 мДж/cм2,
Ptot
=712 Toрр,
PO2 =182 Toрр,
T=300 K для нескольких значений давлений CO2 .
3) Эффект замедления скорости восстановления озона. Величина скорости восстановления озона является одним из ключевых в моделировании кинетики озона в атмосфере. Мы обнаружили, что в наших экспериментах по УФ фотолизу озона скорость восстановления была намного ниже, чем это предсказывается на основе известных кинетических констант процесcов в кислородсодержащей смеси. Результаты представленные на рис. 5 указывают, что скорость восстановления зависит сильно от состава среды. Для смеси O2/O3/Ar (нижняя кривая) время восстановления tO3 составляет около 50 микросек против ожидаемых 15 микросек. Замещение Ar на CO2 приводит к уменьшению tO3. Для смеси O2/O3/CO2 (верхняя кривая на рис. 5) предсказываемое и наблюдаемое времена восстановления озона практически совпадают. Анализ наших результатов показывает, что эффект замедления скорости восстановления озона обусловлен протеканием процесса (2).
O2(a1Δ) +O2(X3Σ) → 2O2(X3Σ). (10)
Отношение скорости процесса (2) к процессу (10) выражается соотношением

Мы рассчитали значения RΔ (представленные на рис. 6) для высот атмосферы от 60 до 105 км, используя стандартные атмосферные концентрации компонент. Как видно из рисунка скорости сравниваемых процессов практически одного порядка в близи 90 км. Процесс (3) дает основной вклад в стабилизацию O3(υ) на высотах ниже 90 км, а процесс (4) выше, результатом чего максимум концентрации O3(υ) достигается вблизи 90 км.

Рис. 6. Отношение скоростей процессов (2) к (10) для высот атмосферы в пределах 65-105 км.
![]()
Как иллюстрируется рисунком 7 на высотах меньших
80 км наработанные в рекомбинационном процессе молекулы O3(υ) в основном успевают стабилизироваться в радиационном (6) и столкновительном
(3) процессах. Для высот H≥80 км вклад в процесса (3) в стабилизацию незначителен.
Здесь O3(υ)
термостабилизируется в процессах (4b) и (6). Химический процесс (4a)
приводит к существенным потерям O3(υ) до 50% на высотах вблизи 100 км.
Следует
отметить, что скорость процесса (4a) даже сравнима со скоростью фотодиссоциации озона при H≥80 км в дневное время суток.

Рис. 7. Доля потерь колебательно-возбужденного озона в процессах (2) и (4a)
в зависимости от высоты атмосферы.
Колебательно-возбужденный озон реагирует с высокой эффективностью с O2(a1Δ) и О в кислородсодержащей смеси. Эти реагенты являются ключевыми компонентами атмосферы и кислородсодержащей плазмы, поэтому процессы (2) и (4) необходимо брать в расчет при моделировании концентрационных профилей этих компонент включая и озона в этих средах.
Процессы (2) и (4a) могут давать заметный вклад в ошибку измерений кинетических констант процессов в среде O/O2/O3. Процесс (2) приводит к занижению значения константы скорости процесса (1) при его измерениях, тогда как процесс (4a) к завышению.
Получение синглетного кислорода в реакции
O(1D)
+ N2O.
Реакция O(1D) с N2O имеет три канала продуктов
→ N2 + O2(a1Δ,X3Σ) (b)
→ O(3P) + N2O (c)
Она интенсивно изучалась, так как один из продуктов реакции NO играет важную роль в разрушении озонового слоя. База данных по атмосферной химии IUPAC рекомендует следующие относительные выходы каналов продуктов реакции: (a) - 0.62, (b)- 0.38 и c < 0.0086. Закон сохранения спина и ab initio расчеты (Gonzales et al. J. Chem. Phys., 115, 2001, 7015, doi: 10.1063/1.1398101), показывают, что синглетный кислород должен образовываться в канале (b), но никаких экспериментальных подтверждений этому получено не было.
Мы провели экспериментальные исследования данного процесса. Излучение эксимерного ArF лазера на длине волны 193 нм использовалось для фотолиза N2O с целью получения возбужденных кислородных атомов O(1D). Кинетика O2(a1Δ) отслеживалась по наблюдению за его люминесценцией на 1268 нм. Калибровка системы измерения O2(a1Δ) осуществлялась с использованием хорошо изученного процесса - фотолиза озона на длине волны 248 нм.
O3+ 248 нм → O2(a1Δ)+ O(1D)
→ O2(X3Σ)+ O(3P)
Излучение кислорода на переходе O2 a-X пропускалось через монохроматор, который выделял нужный переход на длине волны 1268 нм с полосой пропускания по полувысоте 30 нм. Экспериментальная кривая обозначенная как I(O3) на рис. 8 показывает типичную время-разрешенную зависимость интенсивности излучения O2(a1Δ) в экспериментах с фотолизом озона при общем давлении P(tot)=775 Toрр, парциальном давлении озона P(O3)=1.3 Toрр. Уровень сигнала сразу после фотолизного импульса пропорциональна концентрации O2(a1Δ).
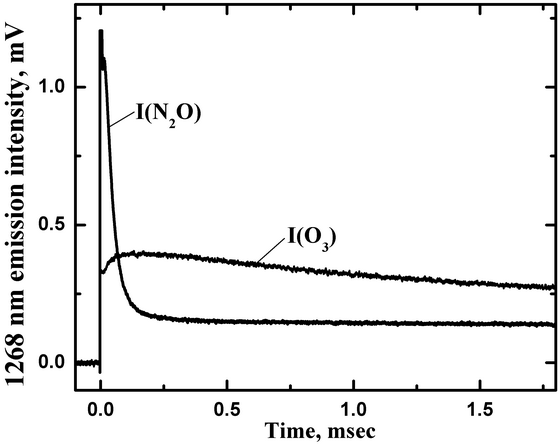
Рис. 8. Временные профили интенсивностей излучения на длине волны 1268 нм для фотолиза озона (I(O3)) при P(N2)=773.7 Toрр, P(O3)=1.3 Toрр, и фотолизе N2O (I(N2O)) при P(N2O)=207 Toрр и P(Ar)=407 Toрр.
Ar был использован в качестве разбавителя N2O, так как он слабо тушит O(1D) (5x10-13 cм3/с) по сравнению с N2O (1.2x10-10 cм3/с). Долгоживущее излучение на 1268 нм также наблюдалось в экспериментах по фотолизу N2O на длине волны 193 нм, подтверждая образование O2(a1Δ). Кривая на рис. 8, обозначенная как I(N2O), показывает пример время-разрешенного сигнала на 1268 нм после импульсного фотолиза смеси N2O/Ar при P(N2O)=207 Toрр и P(Ar)=407 Toрр. Часть кривой с быстрым спадом на начальном участке (0-0.2 мс) принадлежит излучению возбужденного NO2 образованного в реакции
O(3P)+NO+Ar → NO2*+Ar
NO образуется в канале (a), а атомы O(3P) в канале (c). Из-за громадного различия во временах радиационного излучения (на фактор 108) небольшое количество NO2* способно породить излучение сравнимое с излучением концентрированного O2(a1Δ). К счастью процесс наработки NO2* заканчивается намного раньше, чем успевает происходить существенное тушение синглетного кислорода.
Было найдено, что молекулы O2, образованные в канале (b), все электронно-возбужденные O2(a1Δ). Таким образом, относительный выход канала продуктов реакции (b) (0.38) также является величиной выхода для наработки O2(a1Δ) (Azyazov V.N. et al., J. Phys. Chem. A., 2007, vol. 111, p. 6592-6599, doi: 10.1021/jp066531c). Наработка O2(a1Δ) в канале (b) может дать вклад в свечение атмосферы в ближней ИК области спектра. УФ фотолиз N2O удобный источник синглетного кислорода в лабораторных исследованиях, как это было нами продемонстрировано (Azyazov V.N., Heaven M.C., Chem. Phys. Lett., vol. 502, 2011, pp. 150-153, doi: 10.1016/j.cplett.2010.12.053).
Основные результаты:
1. Обнаружен эффект быстрого тушения синглетного кислорода в чистой кислородно-озоновой среде (Azyazov V.N. et al. Chem. Phys. Lett., vol. 482, 2009, pp. 56-61, doi: 10.1016/j.cplett.2009.09.095).
2. Обнаружен эффект неполного восстановления озона в чистой кислородно-озоновой среде (Azyazov V.N. and Heaven M.C., 68th Intern. Symp. on Molecular Spectroscopy, 17-21 June 2013, Columbus)
3. Обнаружен эффект замедления скорости восстановления озона в чистой кислородно-озоновой среде (Azyazov V.N. et al. 32nd Intern. Symp. on Free Radicals, 21-26 July 2013, Potsdam, Germany).
4. Измерен относительный выход синглетного кислорода в процессе O(1D) + N2O → O2(a1Δ) + N2 составляющего > 0.9) (Azyazov V.N. et al., J. Phys. Chem. A., 2007, vol. 111, p. 6592-6599, doi: 10.1021/jp066531c)